🐞 Diagnosing a bug when we were running tximport
Refine.bio has now more than 1MM pre-processed samples readily available for anyone to download, we hope this to help as many scientists as possible. With the system we built, we have been able to process around 46% of all the samples we have discovered. This is mostly due to a lack of standardization in how these samples are uploaded to publicly funded repositories.
Ideally, we would like to process as many of these samples as possible, however, this might be hard given the limited engineering resources that we have. In this post I track down one bug that was making several samples fail, it took me about a day just to detect what was going wrong.
Some Context
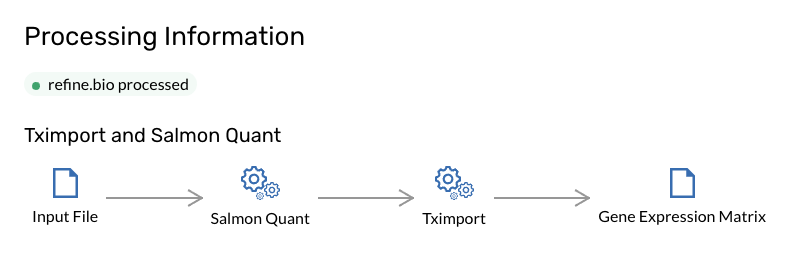
All samples have one of two technologies, they can either be Microarray or Rna-Seq samples. To process the Rna-seq samples, we first apply Salmon and then tximport to process all the samples. This is explained in detail in the refinebio docs.
The short version is that for each experiment, Salmon is applied to every sample, and this produces
quant.sf files. Then we take all of these files together and run tximport on them.
With very shallow knowledge of all this, I started investigating one bug that we were having where tximport was failing. It all started with the following error:
select id, failure_reason from processor_jobs where pipeline_applied='TXIMPORT' and created_at > '2019-08-12 00:00:00'::timestamp and success='f' and failure_reason is not null order by end_time desc limit 1 OFFSET 4;
id | failure_reason
---------+---------------------------------------------------------------------------------------------------------------------------------
3374939 | Found non-zero exit code from R code while running tximport.R: Read 1658 items +
| Parsed with column specification: +
| cols( +
| gene_id = col_character(), +
| tx_name = col_character() +
| ) +
| reading in files with read_tsv +
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 Error: all(txId == raw[[txIdCol]]) is not TRUE+
| In addition: Warning message: +
| In txId == raw[[txIdCol]] : +
| longer object length is not a multiple of shorter object length +
| Execution halted
(1 row)
Find the bug
The error seems very cryptic, it’s something in R that’s triggering it. I thought of first starting to investigate the experiments that we were trying to process where the error was triggered.
select samples.id, samples.accession_code
from samples, original_file_sample_associations, processorjob_originalfile_associations
where samples.id=original_file_sample_associations.sample_id
and original_file_sample_associations.original_file_id=processorjob_originalfile_associations.original_file_id
and processorjob_originalfile_associations.processor_job_id=3374939;
id | accession_code
--------+----------------
415497 | SRR1954375
(1 row)
select experiments.id, accession_code, num_total_samples, title
from experiments inner join experiment_sample_associations on experiments.id=experiment_sample_associations.experiment_id
where experiment_sample_associations.sample_id=415497;
id | accession_code | num_total_samples | title
-------+----------------+-------------------+-----------------------------------------
10884 | SRP056902 | 1659 | Single-cell RNA-seq of murine epidermis
(1 row)
We have a page for this experiment on refine.bio https://www.refine.bio/experiments/SRP056902/single-cell-rna-seq-of-murine-epidermis.
First theory
The first theory we had is that some samples could be associated with multiple experiments, and this somehow could be causing the bug because tximport could be picking up the wrong files. This is not the first data integrity issue that we have had.
select count(S.id), experiment_sample_associations.experiment_id
from samples as S inner join experiment_sample_associations on S.id=experiment_sample_associations.sample_id
where
S.id in (select sample_id from experiment_sample_associations where experiment_id=10884)
group by experiment_sample_associations.experiment_id;
count | experiment_id
-------+---------------
1659 | 10884
(1 row)
With that, we disprove our theory, seems like all 1659 samples belong to a single experiment.
What else?
Second theory
When I’m tracking down a bug, I usually start to get anxious if I run out of theories of what might be wrong. If we have reasons why the program might be failing, we can create an action plan to move forward.
Another thing that could be wrong are the quant.sf files that Salmon is generating, remember
we feed all of these to tximport to process a full experiment.
If we look at the original error:
Error: all(txId == raw[[txIdCol]]) is not TRUE+
Seems like some of the values in txId doesn’t match raw[[txIdCol]], in the original tximport code
that raw[[txIdCol]] is a column in the quant files, and they check that this column matches for all files that are
passed into tximport. There’s probably one file where the order of that column is different.
It might be worth to look into the logs to see if there’s any additional information:
2019-08-13 15:38:09,333 i-02a4007a990254e8b [volume: 1] rkers.processors.salmon ERROR [processor_job: 3374939] [ ... ]] [experiment: 10884] [quant_file_paths: { ... }] [cmd_tokens: ['/usr/bin/Rscript', '--vanilla', '/home/user/rkers/processors/tximport.R', '--file_list', '/home/user/data_store/processor_job_3374939/tximport_inputs.txt', '--gene2txmap', '/home/user/data_store/TRANSCRIPTOME_INDEX/MUS_MUSCULUS/short/genes_to_transcripts.txt', '--rds_file', '/home/user/data_store/processor_job_3374939/txi_out.RDS', '--tpm_file', '/home/user/data_store/processor_job_3374939/gene_lengthScaledTPM.tsv']]: Found non-zero exit code from R code while running tximport.R: Read 1658 items
2019-08-13 15:38:09,577 i-02a4007a990254e8b [volume: 1] rkers.processors.utils ERROR [processor_job: 3374939] [failure_reason: Found non-zero exit code from R code while running tximport.R: Read 1658 items
2019-08-13 15:38:17,602 i-02a4007a990254e8b [volume: 1] rkers.processors.utils ERROR [no_retry: False] [processor_job: 3374939] [pipeline_applied: TXIMPORT] [failure_reason: Found non-zero exit code from R code while running tximport.R: Read 1658 items
It’s not much more, these are pretty much the same error that we already had and that was logged by the processor job.
So, let’s get ALL the computed files to see if they have some column that doesn’t match.
select samples.id, samples.accession_code, computed_files.s3_bucket, computed_files.s3_key
from samples
inner join sample_computed_file_associations on samples.id=sample_computed_file_associations.sample_id
inner join computed_files on computed_files.id=sample_computed_file_associations.computed_file_id
where
samples.id in (select sample_id from experiment_sample_associations where experiment_id=10884)
order by samples.accession_code;
id | accession_code | s3_bucket | s3_key
--------+----------------+--------------------------------+----------------------------------------------------
352851 | SRR1953209 | data-refinery-s3-circleci-prod | w9c27kpc5p10u86giidctyg2_salmontools-result.tar.gz
352851 | SRR1953209 | data-refinery-s3-circleci-prod | eqnu9evrtenl8qihjz16ev2j_multiqc_data.zip
352918 | SRR1953210 | data-refinery-s3-circleci-prod | su98m372y773veys0desxpr0_salmontools-result.tar.gz
353008 | SRR1953211 | data-refinery-s3-circleci-prod | 9go7vfo0l9qb9ldohyefpyb3_salmontools-result.tar.gz
353114 | SRR1953212 | data-refinery-s3-circleci-prod | ip32mdp0prk0c467bbvh9v75_salmontools-result.tar.gz
353223 | SRR1953213 | data-refinery-s3-circleci-prod | xu1wsgs6dvsvhblh6zyqpn3r_multiqc_data.zip
353334 | SRR1953214 | data-refinery-s3-circleci-prod | 6lnhwhmrwi9cio21fq6lbb9j_multiqc_data.zip
353453 | SRR1953215 | data-refinery-s3-circleci-prod | ea1onposi8wshet29c1solsn_salmontools-result.tar.gz
[...]
Well, that doesn’t return the actual quant.sf files that we need…
Talking with my colleagues, another theory came up…
Third theory (hopefully last one)
Salmon needs to use an index
to “quantify Rna-seq reads”. We generate this index for each organism before applying Salmon. This could
be a strong reason why the genes in the quant.sf files were different. Also, we recently updated salmon.
The first step would be to confirm if all the samples in our failing experiment were processed with one
or more versions of salmon.
This query might be harder to write, we want to know the salmon version that was used to build the samples in the experiment. We know we can get all the ids of these samples with:
select sample_id from experiment_sample_associations where experiment_id=10884
At this point, I decided to create a diagram of the DB model as it was starting to be hard to keep all these relations in my head.
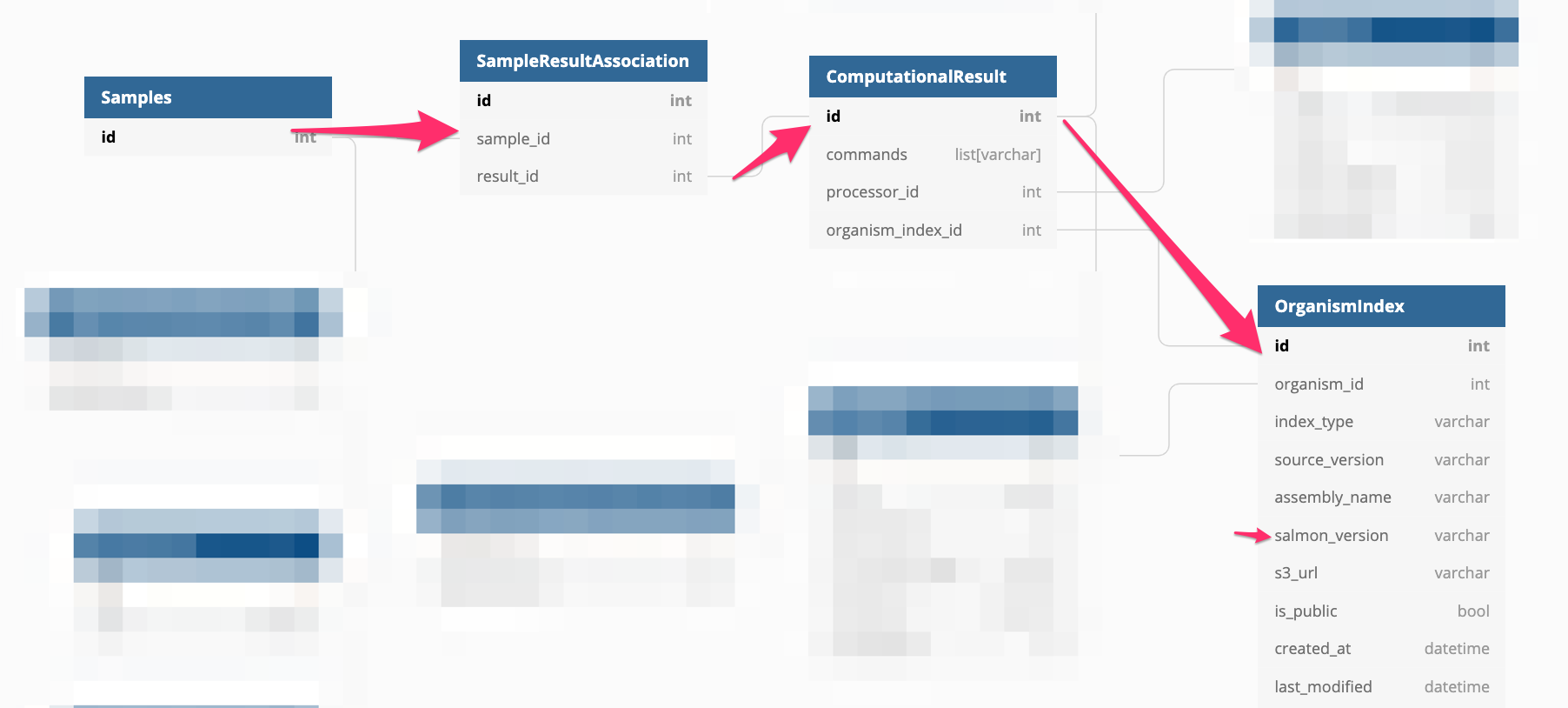
select count(distinct(samples.id)), organism_index.salmon_version
from samples
inner join sample_result_associations on samples.id=sample_result_associations.sample_id
inner join computational_results on sample_result_associations.result_id=computational_results.id
inner join organism_index on computational_results.organism_index_id=organism_index.id
where
samples.id in (select sample_id from experiment_sample_associations where experiment_id=10884)
group by organism_index.salmon_version;
count | salmon_version
-------+----------------
79 | salmon 0.13.1
1579 | salmon 0.9.1
(2 rows)
Finally! some light into this issue. Samples were processed with different versions of Salmon which might cause the gene identifiers in the quant files to be different. It would be a shame to have to re-process all the samples with the old version of salmon. We can look into the files to see if there’s some easy transformation that we can do.
Let’s light a few candles 🕯️🕯️🕯️🕯️🕯️
Quant files
From the code seems that we get the quant files from the computational results associated with the samples. But if samples are also associated with their computed files, maybe we can get them from there more easily?
select samples.id, samples.accession_code, computed_files.s3_bucket, computed_files.s3_key
from samples
inner join sample_computed_file_associations on samples.id=sample_computed_file_associations.sample_id
inner join computed_files on computed_files.id=sample_computed_file_associations.computed_file_id
where
samples.id in (select sample_id from experiment_sample_associations where experiment_id=10884)
and computed_files.filename='quant.sf'
order by samples.accession_code;
id | accession_code | s3_bucket | s3_key
----+----------------+-----------+--------
(0 rows)
They aren’t :/
A colleague mentioned that we don’t link them. Let’s try joining through the computational results.
select samples.id, samples.accession_code, organism_index.salmon_version, computed_files.s3_bucket, computed_files.s3_key
from samples
inner join sample_result_associations on samples.id=sample_result_associations.sample_id
inner join computational_results on sample_result_associations.result_id=computational_results.id
inner join processors on computational_results.processor_id=processors.id
inner join computed_files on computed_files.result_id=computational_results.id
inner join organism_index on computational_results.organism_index_id=organism_index.id
where
samples.id in (select sample_id from experiment_sample_associations where experiment_id=10884)
and processors.name='Salmon Quant'
and computed_files.filename='quant.sf'
and computed_files.s3_key is not null
and computed_files.s3_bucket is not null
order by samples.accession_code;
id | accession_code | salmon_version | s3_bucket | s3_key
--------+----------------+----------------+--------------------------------+------------------------------------
352851 | SRR1953209 | salmon 0.9.1 | data-refinery-s3-circleci-prod | quant_files/sample_352851_quant.sf
352918 | SRR1953210 | salmon 0.9.1 | data-refinery-s3-circleci-prod | quant_files/sample_352918_quant.sf
353008 | SRR1953211 | salmon 0.9.1 | data-refinery-s3-circleci-prod | quant_files/sample_353008_quant.sf
353114 | SRR1953212 | salmon 0.9.1 | data-refinery-s3-circleci-prod | quant_files/sample_353114_quant.sf
353223 | SRR1953213 | salmon 0.9.1 | data-refinery-s3-circleci-prod | quant_files/sample_353223_quant.sf
[...]
360230 | SRR1953246 | salmon 0.13.1 | data-refinery-s3-circleci-prod | quant_files/sample_360230_quant.sf
[...]
That works! Let’s look into the quant files associated with the last two samples and see the differences.
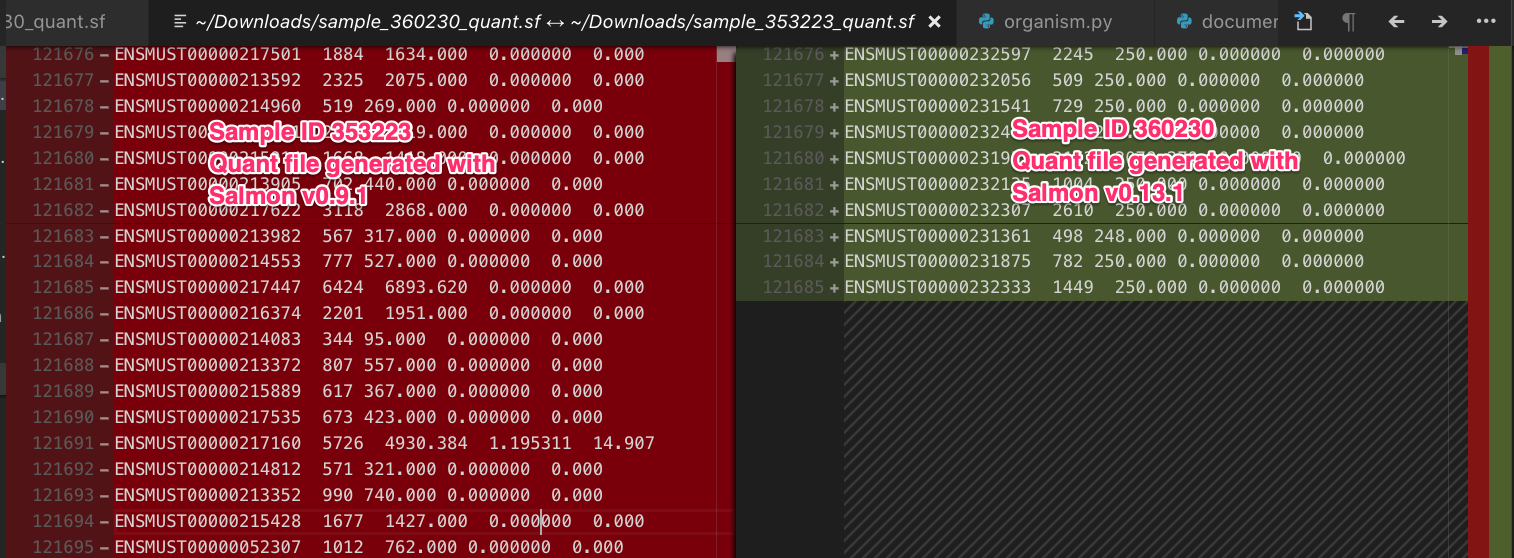
This confirms everything, now we know what caused the issue in the first place, the quant files were generated with different versions of salmon. Initially, we were hoping that the files had at least the same number of rows and that it was just that the ids were in a different order. Since that’s not the case we’ll need to re-run salmon on all samples before running tximport. I filed an issue and started working on the fix.
The fix
Fixing it is easier, once we have the problem diagnosed. I had to add two things for this, first, make sure that tximport is only called if all samples have been processed with the same version of Salmon since we know it will fail otherwise.
We also needed a command to find the experiments were samples had been processed with more than one version of Salmon, and then re-process those. All this was added with this pull request and a further improvement here.
The biggest challenge was trying to optimize various DB queries to make less and make them more efficient. My
a favorite one was using Subquery
to get a Queryset with the computational results associated with an experiment.
newest_computational_results = ComputationalResult.objects.all()\
.filter(
samples=OuterRef('id'),
processor__name=ProcessorEnum.SALMON_QUANT.value['name']
)\
.order_by('-created_at')
computational_results_ids = experiment.samples.all().annotate(
latest_computational_result_id=Subquery(newest_computational_results.values('id')[:1])
)\
.filter(latest_computational_result_id__isnull=False)\
.values_list('latest_computational_result_id', flat=True)
return ComputationalResult.objects.all().filter(
id__in=computational_results_ids
)
Conclusions
Working with a complex database has many challenges. It’s useful to have a diagram of the model when making queries against it. seems like a good option to make them.
When looking for reasons that might be causing a bug, it’s good to have a running list of theories of why the issue is appearing and then go one by one trying to confirm or discard them.